
La diversité des gènes – qui doit être testé et pour quoi?
Vielen Dank für Ihr Interesse!
Einige Inhalte sind aufgrund rechtlicher Bestimmungen nur für registrierte Nutzer bzw. medizinisches Fachpersonal zugänglich.
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
L’augmentation du taux de cholestérol-LDL n’est pas seulement la conséquence du mode de vie personnel: les prédispositions génétiques ont également une influence sur l’existence d’un risque cardiovasculaire accru attribuable à des limitations fonctionnelles du métabolisme des lipides. Quels gènes jouent un rôle à cet égard, qui devrait être testé et ce que révèlent les résultats du test sur le risque cardiovasculaire, tel était l’un des thèmes du premier cours de lipidologie en Suisse en janvier 2024.
Keypoints
-
Les variants pathogènes du LDLR, de l’APOB et du PCSK9 augmentent considérablement le risque CV.
-
L’importance de l’augmentation du risque dépend du gène concerné et du type de variant pathogène.
-
À taux de C-LDL égal, les personnes atteintes de cholestérolémie familiale (HF) ont un risque CV plus élevé que celles qui ne présentent pas de mutation.
-
L’effet des variants pathogènes est modulé par des effets polygéniques (PRS).
-
Les PRS expliquent aussi en grande partie les différences de taux de C-LDL chez les personnes qui ne sont pas atteintes d’HF.
-
Le conseil génétique est important, tant avant qu’après le test.
Le risque cardiovasculaire (CV) est influencé par la variabilité génétique des facteurs participant au métabolisme du cholestérol-LDL (C-LDL), comme l’a expliqué Thomas von Känel, PhD, chef du Service de génétique médicale, Institut central des Hôpitaux, Hôpital du Valais. Les mutations du récepteur des LDL (LDLR), de la proprotéine convertase subtilisine/kexine de type 9 (PCSK9) et de l’apolipoprotéine B (APOB) jouent en particulier un rôle important. «Il en résulte des niveaux sériques élevés de C-LDL et, par conséquent, un risque cardiovasculaire accru.1 Il existe bien sûr d’autres facteurs qui sont également influencés par la génétique, comme des niveaux élevés de lipoprotéine (a) ou l’hypertension artérielle.»
De toutes les variabilités génétiques associées au métabolisme des LDL, qui sont finalement responsables de l’hypercholestérolémie familiale (HF), la mutation du LDLR représente la plus grande part, avec 60 à 80% des cas. Une mutation de perte de fonction, à savoir une mutation d’arrêt ou une grande délétion, entraîne la perte de fonctionnalité du LDLR. Dans 1 à 5% des cas, on trouve des mutations dans l’APOB, où des variants faux-sens donnent naissance à une protéine avec un ligand ApoB dysfonctionnel, et dans jusqu’à 3% des cas, des mutations dans PCSK9, où des variants de gain de fonction renforcent la dégradation du LDLR. «En termes de diagnostic différentiel, des modifications d’autres gènes pertinents peuvent également être envisagées, mais ces trois-là sont les plus fréquentes», a déclaré T.von Känel.
Hypercholestérolémie familiale – le génotype détermine le phénotype
«Dans l’hypercholestérolémie familiale, on distingue en principe, selon le site de la mutation, la forme hétérozygote, HFHe, dans laquelle seul un des deux allèles du LDLR, de l’APOB ou du PCSK9 est affecté, et la forme biallélique (HFHo), dans laquelle les deux allèles présentent une mutation. Si la même mutation est présente sur les deux allèles, on parle d’HF homozygote proprement dite, mais qui est plutôt rare. Si les deux allèles sont affectés par deux mutations différentes, on est en présence de la forme «hétérozygote composée» de l’HF, plus fréquente. Dans de très rares cas, une forme hétérozygote double, dite digénique, est également à l’origine de la maladie: un allèle de deux gènes différents, par exemple le LDLR et l’APOB, est alors affecté», a expliqué T.von Känel pour expliquer la classification.
La forme hétérozygote est présente en Suisse avec une fréquence d’environ 1:250. Le mode de transmission est autosomique dominant, ce qui signifie que les parents au premier degré (parents, enfants) ont un risque de 50% de présenter également la mutation. Sur le plan phénotypique, on constate une forte augmentation du taux de C-LDL et des xanthomes tendineux et cutanés, ainsi qu’un arc cornéen dès l’âge de <45 ans. Les maladies CV apparaissent tôt dans la vie, un homme non traité sur deux présentant déjà un événement CV avant l’âge de 50 ans. Le traitement repose sur les statines, les inhibiteurs de la PCSK9 et à la modification du mode de vie.2,3
La forme biallélique de l’hypercholestérolémie familiale est nettement plus rare, avec une fréquence de 1:400’000 à 1:160’000 en Suisse. L’hérédité est autosomique récessive, ce qui signifie que les parents au premier degré ont certainement une forme hétérozygote d’HF. Sur le plan phénotypique, ils présentent des taux de C-LDL très élevés (>12,9mmol/l), des xanthomes (parfois aussi dans les espaces interdigités) et, généralement, des maladies CV avant l’âge de 30 ans.2,3 «Le risque d’infarctus du myocarde est dramatiquement accru chez ces personnes», a souligné l’expert. «Malheureusement, les statines ne sont généralement pas efficaces dans cette forme de la maladie, car il faudrait pour cela une fonction résiduelle du LDLR, qui n’est pas présente la plupart du temps.»
Selon T.von Känel, l’expression clinique de la maladie dépend toutefois fortement du gène muté et du type de variant pathogène; on constate donc une corrélation génotype-phénotype. «En cas de mutation du LDLR, le risque de maladie coronarienne est assez fortement accru, l’odds ratio est proche de 4 pour tous les variants génétiques, voir même de 7 pour une survenue précoce de la maladie coronarienne.4 On peut encore distinguer s’il s’agit d’une mutation de perte de fonction, pour laquelle le risque est le plus élevé avec un odds ratio d’environ 9, ou d’une mutation faux-sens avec fonction résiduelle, associée à un risque un peu plus faible.»5
Qu’est-ce que cela signifie sur le plan clinique? «Les personnes atteintes d’hypercholestérolémie familiale ont un risque beaucoup plus élevé de maladie coronarienne comparativement à celles qui ne présentent pas de variants génétiques, tout en ayant des taux de C-LDL aussi élevés. Le risque augmente de manière exponentielle avec le niveau de C-LDL»,5 a expliqué l’expert (Fig.1). Le concept d’exposition cumulative au C-LDL permet également d’identifier le risque nettement accru:6 Les personnes présentant une mutation homozygote du LDLR atteignent, selon leur autre profil de risques (sexe masculin, hypertension, diabète, statut tabagique), dès l’adolescence, des valeurs cumulatives qui entraînent un risque CV accru. En cas de mutation hétérozygote, la valeur seuil est atteinte dès le milieu de la vingtaine, alors que les personnes sans mutation n’entrent dans la zone de risque qu’à partir de la cinquantaine en moyenne (Fig.2). «Il faut le savoir et adapter en conséquence le traitement de ces patients et patientes à risque accru. Il faut également savoir qu’il existe une pénétration incomplète dans le phénotype biochimique: alors que la valeur du C-LDL dans le groupe des personnes porteuses d’une mutation du LDLR est en moyenne de 4,9mmol/l, environ 27% d’entre elles présentent tout de même des valeurs <3,4mmol/l – ce qui correspond à la valeur moyenne du C-LDL de celles qui ne présentent pas de mutation.»5
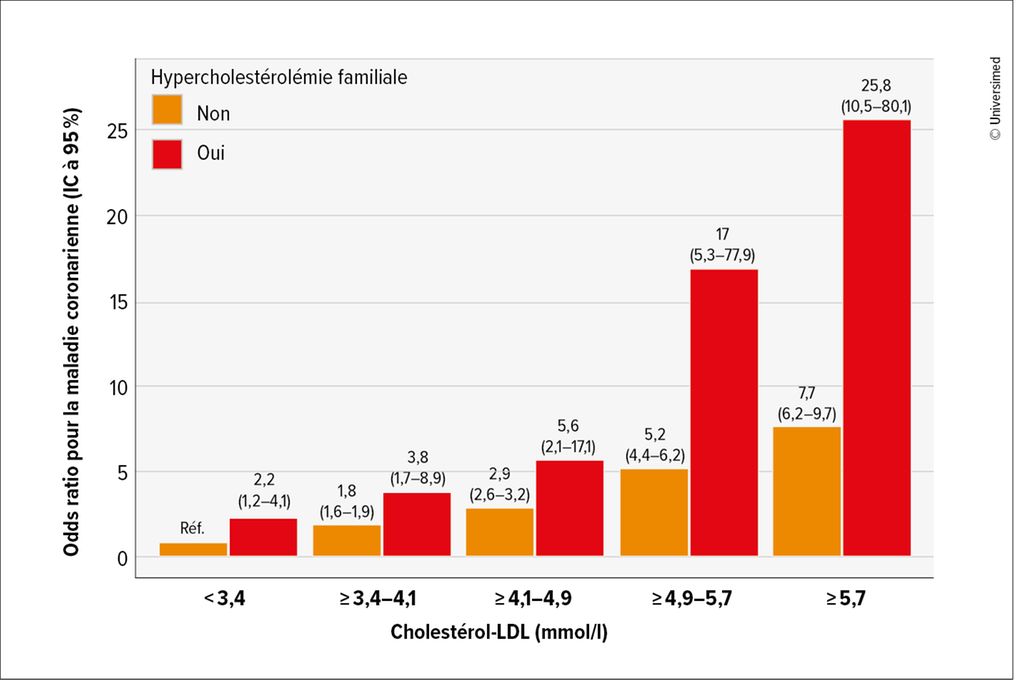
Fig. 1: Risque cardiovasculaire en fonction du taux de C-LDL pour les personnes avec et sans hypercholestérolémie familiale (adaptée de Khera AV et al. 2016)5
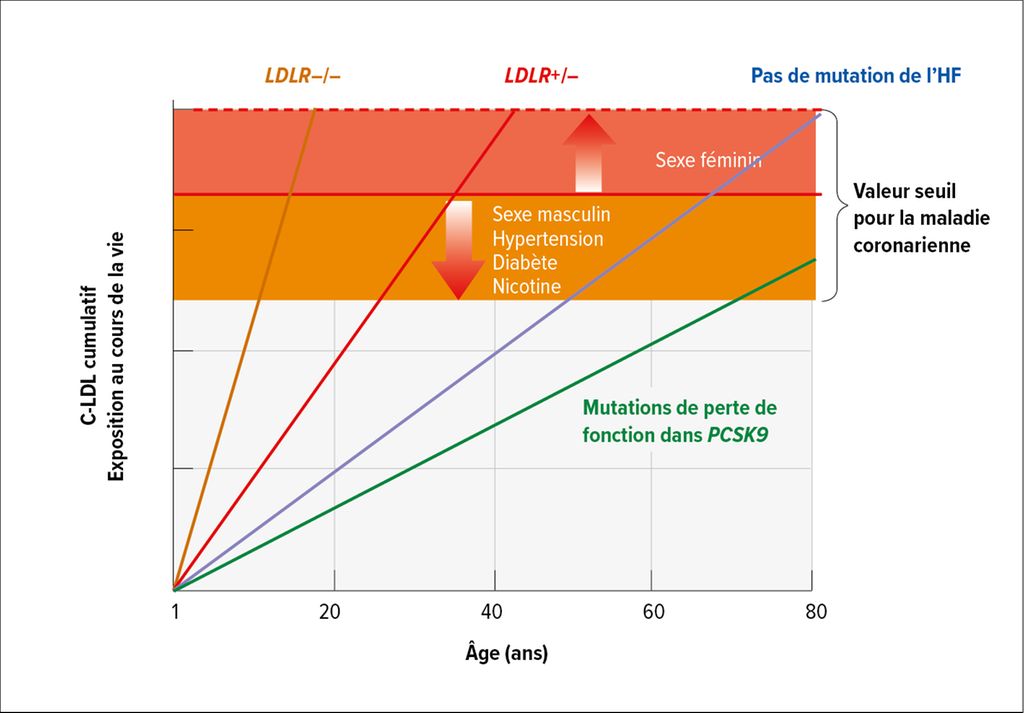
Fig. 2: Exposition cumulative au C-LDL en fonction de l’âge pour les personnes atteintes d’hypercholestérolémie familiale (HF) homozygote (LDLR–/–) ou hétérozygote (LDLR+/–) et les personnes sans mutation d’HF. La ligne rouge représente le seuil théorique d’exposition cumulée au C-LDL nécessaire pour présenter une maladie coronarienne, ce seuil étant plus bas en présence d’autres facteurs de risque cardiovasculaire (adaptée de Snidermann AD et al. 2024)6
Importance des tests génétiques pour l’établissement du diagnostic
Selon les critères du Dutch Lipid Clinic Network (DLCN), le score global tient compte non seulement du taux de C-LDL, mais aussi de l’anamnèse et de l’examen clinique de la personne affectée, des antécédents familiaux et des résultats des tests génétiques concernant une mutation de LDLR, d’APOB et de PCSK9. Ce score peut également être consigné et interprété en ligne via le calculateur HF du GSLA.
Prescription et remboursement
«Lors de la prescription du test génétique, la partie la plus importante est le conseil génétique médical avant le test», a souligné T.von Känel. Il s’agit notamment de recueillir les antécédents médicaux personnels et familiaux et de fournir des informations sur la maladie ainsi que sur les bienfaits et les risques du test – dans le but d’obtenir le consentement (ou le refus) éclairé de la personne affectée. Les documents nécessaires comprennent la déclaration de consentement, le formulaire de commande signé ainsi que la garantie de prise en charge des frais par la caisse-maladie (voir encadré «Liens utiles»). Ces documents sont envoyés au laboratoire avec le matériel d’analyse – un grand tube de sang EDTA – qui est ensuite transmis au laboratoire spécialisé correspondant. «En soi, tout est très simple – sauf en ce qui concerne la prise en charge des frais. En effet, l’HF ne figure pas sur la liste officielle des analyses et la position ‹maladie orpheline› n’est applicable qu’aux maladies dont la fréquence est inférieure à 1:2000 – or l’HF se produit avec une fréquence de 1:250. Notre première demande d’inclusion de l’HF comme position explicite dans la liste des analyses a malheureusement été rejetée en 2020.»
Procédure de test
En ce qui concerne le test génétique lui-même, il existe deux variantes: la méthode de séquençage de nouvelle génération (NGS, «next generation sequencing») d’au moins LDLR, APOB et PCSK (CHF 2610.–) convient aux cas index dont le variant pathogène familial n’est pas connu, tandis que le séquençage Sanger (CHF 193.–) peut être utilisé pour les parents du cas index lorsque le variant recherché est déjà connu. Il existe deux approches différentes pour le NGS: le test d’HF ciblé et le séquençage de l’exome entier (WES, «whole exome sequencing»). «Dans le test d’HF ciblé, on ne séquence que les gènes d’intérêt, ce qui limite les coûts et le travail et présente une bonne sensibilité, mais l’inconvénient est qu’on ne peut pas analyser d’autres gènes. Dans le cas du WES, par contre, tous les exons des quelque 20000 gènes sont séquencés, mais seuls les gènes de l’HF sont analysés. Cela représente certes un gros effort et des coûts beaucoup plus élevés, mais on a ainsi la possibilité de pouvoir analyser plus tard d’autres gènes qui deviennent pertinents au fil de l’évolution – par exemple aussi pour un diagnostic de tumeur.»
Interprétation des résultats des tests
L’interprétation des résultats du NGS se fait en fonction de 5 classes de variants – basées sur le type de mutation, la fréquence dans la population générale, les données issues des programmes de prédiction, des bases de données et de la littérature, ainsi que des tests fonctionnels et des connaissances sur la co-ségrégation. Les résultats avec une probabilité de pathogénicité de <1% (classe 1, bénigne) et de <10% (classe 2, probablement bénigne) sont considérés cliniquement comme des résultats négatifs, tout comme les résultats de la classe 3 (variante avec signification incertaine) avec une probabilité de pathogénicité de 10 à 90%. Une probabilité de pathogénicité >90% (classe 4, probablement pathogène) et >99% (classe 5, pathogène) est considérée comme un résultat positif sur le plan clinique.7 «Alors qu’il y a longtemps eu des divergences dans l’interprétation de la pathogénicité des résultats, l’application des critères est désormais plus claire et les résultats entre les différents laboratoires sont congruents depuis la publication des directives de consensus pour la classification des variants du LDLR par un panel d’experts8», explique T.von Känel. «Ce qui est très important, c’est que les résultats doivent être communiqués à la personne concernée dans le cadre d’une consultation génétique. Le conseil génétique se situe donc au début et à la fin d’un test génétique.»
Effets polygéniques – Modificateur du risque
Alors que l’HF repose sur des variants rares ayant des effets importants, l’hypercholestérolémie polygénique, qui peut être déterminée à l’aide du score de risque polygénique, repose sur l’effet cumulatif de différents polymorphismes génétiques influençant le métabolisme du C-LDL, qui – chacun pris séparément – ont un impact faible avec des pertes fonctionnelles à peine perceptibles, mais qui, ensemble, ont un effet clinique.9 Ainsi, des personnes sans mutations monogéniques dans les gènes de l’HF peuvent également présenter un risque CV accru par l’interaction de nombreux polymorphismes individuels avec de faibles effets individuels dans l’ensemble, ou l’effet d’une mutation monogénique peut être modifié par la présence d’effets polygéniques supplémentaires10 – ce qui explique le risque CV plus ou moins élevé chez les personnes atteintes d’HF porteuses de la même mutation monogénique, mais aussi dans la population des personnes non touchées par l’HF. «L’évaluation du risque réel d’une personne repose donc sur le risque clinique, mesuré à l’aide du taux de C-LDL et d’autres facteurs de risque comme le statut tabagique et l’hypertension, plus le risque polygénique, et représente donc un risque combiné», a déclaré T.von Känel pour expliquer l’approche très personnalisée de la détermination du risque. «Malgré tout, certaines questions restent en suspens: quelle est la fiabilité des estimations de risque mentionnées pour d’autres ethnies non européennes? Combien de polymorphismes génétiques faut-il tester pour obtenir des résultats fiables? Est-ce que tout cela a une utilité clinique ou le ‹number to test› pour identifier les personnes à risque élevé est-il tout simplement trop important? En cas de risque polygénique ne suivant pas les lois de l’hérédité de Mendel, dois-je tester les parents des personnes affectées? Et puis des questions très pratiques: qui prescrit le test, qui le paie et qui communique les résultats?», a résumé T.von Känel au sujet des questions en suspens au sujet du diagnostic génétique en lipidologie.
Liens utiles
Calculateur HF du GSLA/Score DLCN
Consentement pour les analyses génétiques
Demande de facturation sous une position de maladie orpheline de la liste des analyses
Source:
1er cours GSLA en lipidologie clinique, 18–19 janvier 2024, Zurich
Littérature:
1 Nordestgaard BG et al.: Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J 2013; 34: 3478-90a 2 Adam MP et al. (eds.) GeneReviews®. Seattle (WA): University of Washington, Seattle; 1993–2024. https://www.ncbi.nlm.nih.gov/books/NBK1116/ 3 Brun N et al.: [New guidelines for screening and management of familial dyslipidemia.] Rev Med Suisse 2016; 12: 435-9 4 Abul-Husn NS et al.: Genetic identification of familial hypercholesterolemia within a single U.S. health care system. Science 2016; 354: aaf7000 5 Khera AV et al.: Diagnostic yield and clinical utility of sequencing familial hypercholesterolemia genes in patients with severe hypercholesterolemia. J Am Coll Cardiol 2016; 67: 2578-89 6 Snidermann AD et al.: The severe hypercholesterolemia phenotype: clinical diagnosis, management, and emerging therapies. J Am Coll Cardiol 2014; 63: 1935-47 7 Richards S et al.: Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17: 405-24 8 Chora JR et al.: The Clinical Genome Resource (ClinGen) Familial Hypercholesterolemia Variant Curation Expert Panel consensus guidelines for LDLR variant classification. Genet Med 2022; 24: 293-306 9 Talmud PJ et al.: Use of low-density lipoprotein cholesterol gene score to distinguish patients with polygenic and monogenic familial hypercholesterolaemia: a case-control study. Lancet 2013; 381: 1293-301 10Fahed AC et al.: Polygenic background modifies penetrance of monogenic variants for tier 1 genomic conditions. Nat Commun 2020; 11: 3635