
Nouvelles approches thérapeutiques et développement des concepts existants
Sie sind bereits registriert?
Loggen Sie sich mit Ihrem Universimed-Benutzerkonto ein:
Sie sind noch nicht registriert?
Registrieren Sie sich jetzt kostenlos auf universimed.com und erhalten Sie Zugang zu allen Artikeln, bewerten Sie Inhalte und speichern Sie interessante Beiträge in Ihrem persönlichen Bereich
zum späteren Lesen. Ihre Registrierung ist für alle Unversimed-Portale gültig. (inkl. allgemeineplus.at & med-Diplom.at)
Le 30e congrès annuel de la World Muscle Society s’est tenu à Vienne du 7 au 11 octobre 2025. Outre les dernières études et données, l’accent a également été mis sur les échanges personnels entre les quelque 15000 participant·es issu·es de 80 pays différents. C’est surtout dans les domaines thérapeutiques de l’amyotrophie spinale, de la dystrophie musculaire de Duchenne et de la dystrophie myotonique que des résultats d’études prometteurs ont été présentés, susceptibles d’améliorer et d’élargir les concepts thérapeutiques existants.
Pendant longtemps, en dehors des corticostéroïdes, les patient·es atteint·es de maladies musculaires disposaient de peu d’options thérapeutiques.
Ces dernières années, l’arrivée de thérapies ciblées a fortement dynamisé ce domaine. L’automne dernier, le congrès de la World Muscle Society (WMS) à Vienne a présenté plusieurs temps forts susceptibles d’apporter de l’espoir aux patient·es souffrant de maladies musculaires comme la dystrophie musculaire de Duchenne.
Mise à jour: amyotrophie spinale
Données convaincantes à 3 ans sur le risdiplam chez des nourrissons atteint·es de SMA présymptomatique
Dans l’amyotrophie spinale (SMA), l’initiation précoce du traitement est déterminante pour le succès thérapeutique; toutefois, la dégénérescence des motoneurones débute déjà avant l’apparition des premiers symptômes.1 La maladie est causée par une mutation du gène «survival motor neuron-1» (SMN1). Le risdiplam constitue l’un des standards thérapeutiques. Ce médicament modifie l’épissage de l’ARNm SMN2, normalement non fonctionnel mais potentiellement compensatoire, permettant ainsi la production d’une protéine SMN fonctionnelle.2,3
Les patient·es atteint·es de SMA en bas âge sont souvent incapables de s’asseoir ou de se retourner sans assistance. «L’étude RAINBOWFISH est une étude de phaseII, à un seul bras, visant à évaluer l’efficacité, la sécurité, la pharmacocinétique et la pharmacodynamie du risdiplam chez des nourrissons (de la naissance à 6 semaines, n=25) présentant une SMA présymptomatique confirmée génétiquement», a expliqué la Prof. Maria Mazurkiewicz-Bełdzinska, de l’Université de médecine de Gdánsk (Pologne).4 Le critère d’évaluation primaire de l’étude a été atteint dès 12 mois: 80% des patient·es présentant deux copies du gène SMN2 ont pu s’asseoir sans soutien pendant plus de 5 secondes.5
Atteinte et maintien des étapes motrices sur 3 ans sous risdiplam
Les données actuelles à 3 ans ont montré que 91% des participant·es ont atteint et conservé les principales étapes motrices, telles que la position assise sans soutien, la station debout et la marche. Ils et elles ont également conservé leur fonction bulbaire et n’ont pas nécessité de soutien respiratoire, en l’absence d’infection.
RAINBOWFISH est la première étude clinique à avoir inclus une évaluation cognitive. Les enfants traité·es par risdiplam pendant 3 ans ont présenté des capacités cognitives comparables à celles d’enfants au développement typique sans SMA. Les résultats étaient similaires dans tous les groupes selon le nombre de copies du gène SMN2. Concernant la sécurité, aucun effet indésirable grave n’a été rapporté au cours des trois années de traitement.4, 5
Étude DEVOTE, partie C: bénéfice thérapeutique sous une dose accrue de nusinersen
«Pour un succès thérapeutique optimal, le traitement de la SMA par le nusinersen devrait être initié le plus tôt possible, en particulier chez les nourrissons et les jeunes enfants», a introduit dans son exposé le Prof. Dr méd. Eugenio Mercuri, de l’Université catholique de Rome (Italie).6
L’étude DEVOTE, une étude de phaseII/III en trois parties, a évalué le bénéfice et le profil de sécurité d’une dose plus élevée de nusinersen (50/28mg).7 La partie C de l’étude a inclus des patient·es (n=40) atteint·es de SMA d’apparition infantile ou tardive, qui avaient reçu nusinersen à la dose 12/12mg pendant ≥1 an avant l’inclusion. Au total, 40% des participant·es à l’étude étaient âgé·es de moins de 18 ans et 60% de plus de 18 ans; ils et elles avaient été traité·es par 12/12mg de nusinersen pendant une durée médiane de 3,9 ans. Les participant·es ont reçu une dose initiale de 50mg quatre mois ±14 jours après leur dernière dose de 12mg, suivie de deux doses d’entretien de 28mg à intervalles de 4 mois.8
Améliorations motrices sous nusinersen 50/28g
Après le passage à une dose de nusinersen de 50/28mg, les participant·es ont présenté, au jour 302, une amélioration des fonctions motrices (Fig.1): l’augmentation moyenne par rapport à la valeur initiale était de +1,8 point sur la Hammersmith Functional Motor Scale Expanded (HFMSE) et de +1,2 point sur le Revised Upper Limb Module (RULM). Ces améliorations sont survenues durant une période au cours de laquelle les bénéfices thérapeutiques obtenus avec la dose standard de 12/12mg évoluent généralement vers un plateau après des améliorations initiales marquées. Le nusinersen 50/28mg a été globalement bien toléré, les signaux de sécurité correspondant largement au profil observé avec la dose de 12/12mg.8 Les résultats de l’étude plaident ainsi en faveur d’un passage d’une dose de 12/12mg à une dose de 50/28mg.
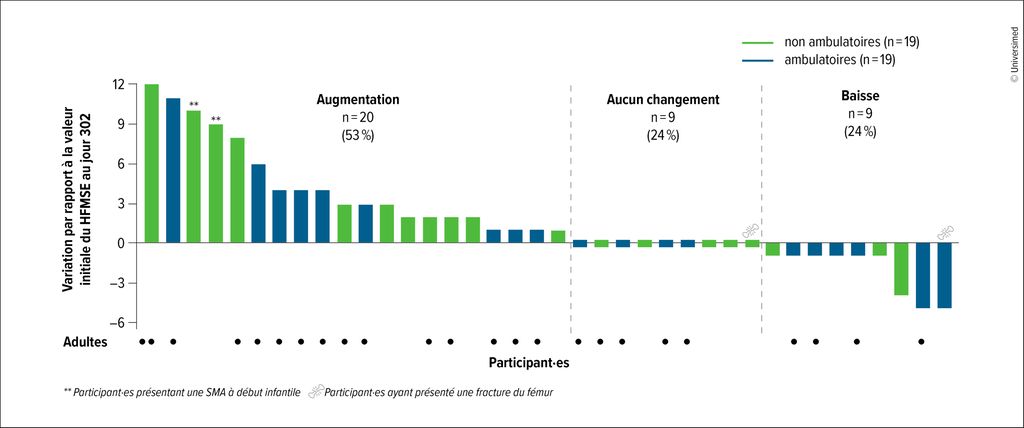
Fig. 1: 53% des participant·es à l’étude DEVOTE ont présenté une amélioration de la fonction motrice sous une dose accrue de nusinersen. Les améliorations ont été évaluées à l’aide de la Hammersmith Functional Motor Scale Expanded (HFMSE) après 302 jours de traitement par nusinersen 50/28mg7
Mise à jour: dystrophie musculaire de Duchenne (DMD)
La DMD est due à une mutation du gène de la dystrophie, transmis selon un mode récessif lié au chromosome X. La protéine dystrophine joue un rôle essentiel en tant que protéine structurelle des muscles squelettiques. La maladie touche donc presque exclusivement les garçons.
Taux de dystrophine insuffisant sous le facteur de saut d’exon 51 PGN-EDO51
Le Prof. Dr méd. Hugh McMillan, du Children’s Hospital of Eastern Ontario à Ottawa (Canada), a présenté les données de l’étude de phaseII CONNECT1-EDO51. Cette étude a évalué l’efficacité du PGN-EDO51, un agent permettant le saut d’exon 51 et, par conséquent, la production d’une dystrophine tronquée mais fonctionnelle. La technologie «Enhanced delivery oligonucleotide» (EDO) optimise l’apport tissulaire ainsi que l’absorption nucléaire de la substance active.9 Des études non cliniques avaient préalablement montré qu’une seule dose de PGN-EDO51 pouvait déjà induire des taux élevés de saut d’exon et une augmentation des niveaux de dystrophine.10
Dans l’étude CONNECT1-EDO51, des patient·es atteint·es de DMD ont reçu quatre doses de PGN-EDO51 sur une période de 3 mois, à une concentration de 5mg/kg (n=3) ou 10mg/kg (n=4). Le profil de sécurité observé était favorable: aucun effet indésirable grave n’a été rapporté sous la dose de 5mg/kg. Sous la dose de 10mg/kg, une hypomagnésémie asymptomatique a été observée chez 2 participant·es. Jusqu’à la semaine 13, l’ensemble des participant·es a présenté une augmentation du saut d’exon, avec un taux médian de 4,26% ainsi qu’une production moyenne de dystrophine de 3,11% contre une valeur initiale de 0,47%.9
Cette exposition inattendue et la faible production de dystrophine suggéraient toutefois que des doses nettement plus élevées seraient nécessaires pour atteindre des niveaux de dystrophine cliniquement pertinents. L’entreprise a par conséquent interrompu le développement du PGN-EDO51 dans l’indication de la DMD.
FORWARD-53: amélioration de la fonction musculaire grâce au saut d’exon 53
Le WVE-N531 est un oligonucléotide à saut d’exon 53 doté d’un squelette modifié, promettant une stabilité accrue, une efficacité renforcée et une meilleure disponibilité tissulaire. L’étude FORWARD-53, une étude de phaseII à un seul bras, a évalué la sécurité et les résultats fonctionnels sous WVE-N531. Li-Jung Tai, de Wave Life Sciences à Cambridge (États-Unis), a présenté la partie B de l’étude, dans laquelle 11 patient·es âgé·es de 5 à 11 ans (dont 10 ambulant·es) ont reçu une dose de WVE-N531 de 10mg/kg toutes les deux semaines pendant 48 semaines.
Le WVE-N531 s’est révélé sûr et bien toléré: l’ensemble des effets indésirables liés au traitement était de sévérité légère à modérée. L’expression de la dystrophine mesurée par Western Blot s’est stabilisée entre la 24e et la 48e semaine de traitement. La valeur moyenne observée était de 7,8%. Au total, 88% des patient·es ont atteint un taux moyen de dystrophine supérieur à 5%, et le taux moyen de saut d’exon s’élevait à 54%. Ces données robustes ont également été confirmées par des analyses histologiques. Après 48 semaines, une amélioration de l’architecture musculaire, un passage de la régénération musculaire à la maturation des fibres musculaires, ainsi qu’une réduction de 28,6% de la fibrose musculaire ont été observés (p<0,01).11
Améliorations cliniques de la fonction musculaire sous WVE-N531
Sous WVE-N531, des différences cliniquement significatives dans l’évolution de la maladie ont également été observées pour plusieurs évaluations fonctionnelles, notamment le «time to rise» (TTR) et le North Star Ambulatory Assessment (NSAA). Ainsi, le TTR s’est amélioré en moyenne de 3,8 secondes par rapport à l’évolution naturelle (p<0,05). Les 11 patient·es ont tous·tes intégré la phase d’extension de l’étude et reçoivent désormais des doses mensuelles de WVE-N531.
Ces données systématiquement positives ont constitué la base d’une nouvelle étude de confirmation globale. En outre, une demande auprès de la Food and Drug Administration (FDA) américaine devrait être déposée en 2026 afin de soutenir une autorisation accélérée du WVE-N531 avec une administration mensuelle.11
Premières données prometteuses issues de l’étude INSPIRE-DUCHENNE
Une autre option thérapeutique dans la DMD repose sur l’utilisation de microdystrophines afin d’augmenter les taux de dystrophine fonctionnelle. SGT-003 est une microdystrophine de deuxième génération intégrant les exons 42 à 45 ainsi que, de manière spécifique, le domaine de liaison nNOS. Cette conception vise notamment à améliorer la circulation sanguine, afin de prévenir les ischémies induites par l’activité et les lésions musculaires associées.12 Les modifications supplémentaires de SGT-003 comprennent la capside SLB101, qui cible les intégrines fortement régulées dans les muscles dystrophiques, ainsi qu’un rapport amélioré de capsides pleines à capsides vides, >80% contre <20%.13 L’ensemble de ces adaptations conduit à une biodistribution accrue et à une expression renforcée dans les muscles squelettiques et le muscle cardiaque.
Niveaux élevés d’expression de la microdystrophine et amélioration de l’intégrité musculaire
INSPIRE-DUCHENNE est une étude de phaseI/II évaluant la sécurité et l’efficacité d’une administration unique du médicament de thérapie génique SGT-003 (1,0 E 14vg/kg) chez des patient·es pédiatriques atteint·es de DMD. Le Prof. Dr méd. Kevin M. Flanigan, du Nationwide Children’s Hospital à Columbus (États-Unis), a présenté les données de la cohorte 1 (4 à <7 ans, n=9) et de la cohorte 2 (7 à <12 ans, n=6).
À la date de clôture de mars 2025, aucun effet indésirable grave n’avait été observé chez les 15 patient·es inclus·es. Les premières données issues de biopsies musculaires de trois patient·es ayant déjà terminé 90 jours de traitement ont montré des taux de transduction constamment élevés, avec une moyenne de 18,7 pour le nombre de copies de vecteur. L’expression de la microdystrophine atteignait 110% de la valeur normale de la dystrophine, et l’analyse immunohistochimique a mis en évidence 78% de fibres musculaires positives pour la microdystrophine (Fig.2). Par ailleurs, des améliorations ont été observées pour plusieurs biomarqueurs de l’intégrité musculaire. Ainsi, les biomarqueurs de lésions musculaires étaient significativement réduits de 45 à 60%.14
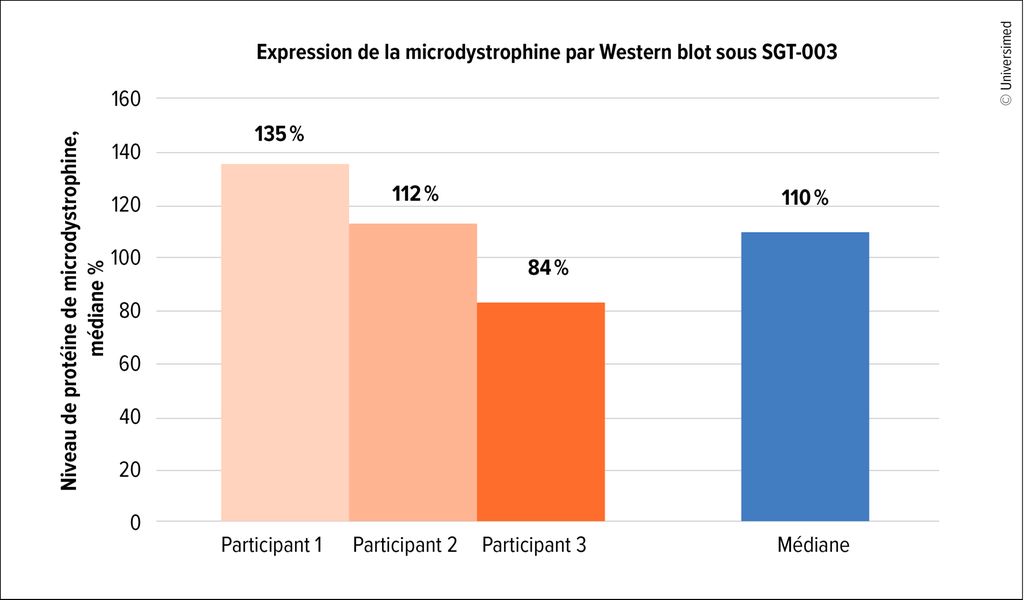
Fig. 2: Les premières données de biopsie musculaires chez des patient·es pédiatriques inclus·es dans l’étude INSPIRE-DUCHENNE ont montré une restauration de l’expression de la microdystrophine par Western blot sous thérapie génique SGT-003
Au vu de ces résultats positifs, l’étude a récemment été étendue et des patient·es plus âgé·es ont été inclus·es dans les cohortes 4 et 5.
Mise à jour: dystrophie myotonique de type 1 (DM1)
La DM1 est une maladie musculaire héréditaire autosomique dominante, caractérisée par expansion de triplets répétés dans le gène de la «dystrophia myotonica protein kinase»(DMPK), entraînant une diminution de la production de la myotonine protéine kinase.
Données initiales prometteuses concernant le PGN-EDODM1 dans la DM1
PGN-EDODM1 est un oligonucléotide conjugué à un peptide (PPMO), reposant sur la technologie peptidique EDO, actuellement évalué dans le contexte de la DM1. Dans la DM1, l’expansion pathogène des répétitions CUG entraîne la formation de structures en épingle à cheveux (hairpins) dans l’ARNm du gène DMPK, lesquelles se lient de manière stable à des facteurs d’épissage tels que le «muscleblind-like splicing regulator 1» (MBNL1). Les anomalies d’épissage qui en résultent sont considérées comme la cause de la maladie.15
«L’effet du PGN-EDODM1 repose sur sa liaison aux répétitions CUG et sur l’augmentation des niveaux de MBNL1 libre obtenue par blocage stérique. Les structures en épingle à cheveux sont en outre résolues, ce qui permet au mécanisme de «proofreading» de fonctionner à nouveau», explique le Prof. Dr méd. Hanns Lochmüller, du Children’s Hospital of Eastern Ontario à Ottawa (Canada). MBNL1 peut alors restaurer les profils d’épissage de plusieurs transcrits en aval, qui constituent une cause centrale de la pathologie de la DM1.16
Taux élevé de correction de l’épissage après une dose unique de PGN-EDODM1
FREEDOM-DM1 est une étude de phaseI contrôlée par placebo menée chez des patient·es atteint·es de DM1. Les participant·es ont reçu une dose unique de PGN-EDODM1 à la concentration de 5mg/kg (n=8), 10mg/kg (n=8) ou 15mg/kg (n=8).
Les premières données cliniques ont montré une augmentation dose-dépendante des concentrations de MBNL1 dans le tissu musculaire. Sous la dose la plus élevée de 15mg/kg, le taux de correction d’épissage le plus élevé jamais mesuré a été atteint, soit 53,7% (Fig.3). De plus, ces corrections de l’épissage ont été observées chez 100% des patient·es. Ces résultats suggèrent que des administrations supplémentaires de PGN-EDODM1 pourraient encore améliorer ces valeurs.

Fig. 3: L’étude de phaseI FREEDOM-DM1 a déjà montré des résultats prometteurs concernant la correction de l’épissage défectueux chez des patient·es atteint·es de dystrophie myotonique de type 1 au jour 28 sous PGN-EDODM1
Par ailleurs, le profil de sécurité du PGN-EDODM1 s’est révélé favorable: aucun effet indésirable grave associé n’a été observé.16 Sur la base de ces données prometteuses, l’étude internationale de phaseII FREEDOM2-DM1 a été initiée afin d’évaluer la sécurité et le bénéfice d’un traitement par doses multiples de PGN-EDODM1.17
Source:
Présentations issues de la «Clinical Trial Updates Session» du congrès WMS, 7–11 octobre 2025, Vienne
Littérature:
1 Govoni A et al.: Time is motor neuron: therapeutic window and its correlation with pathogenetic mechanisms in spinal muscular atrophy. Mol Neurobiol 2018; 55: 6307-18 2 Ratni H et al.: Discovery of risdiplam, a selective survival of motor neuron-2 ( SMN2) gene splicing modifier for the treatment of spinal muscular atrophy (SMA). J Med Chem 2018; 61: 6501-17 3 Poirier A et al.: Risdiplam distributes and increases SMN protein in both the central nervous system and peripheral organs. Pharmacol Res Perspect 2018; 6: e00447 4 An open-label study of risdiplam in infants with genetically diagnosed and presymptomatic spinal muscular atrophy (NCT03779334)5 Finkel RS et al.: Risdiplam in presymptomatic spinal muscular atrophy. N Engl J Med 2025; 393: 671-82 6 Crawford TO et al.: Continued benefit of nusinersen initiated in the presymptomatic stage of spinal muscular atrophy: 5-year update of the NURTURE study. Muscle Nerve 2023; 68(2): 157-70 7 Finkel RS et al.: DEVOTE study exploring higher dose of nusinersen in spinal muscular atrophy: study design and part A results. J Neuromuscul Dis 2023; 10: 813-23 8 Escalating dose and randomized, controlled study of nusinersen (BIIB058) in participants with spinal muscular atrophy(NCT04089566) 9 A phase 2, open-label, multiple ascending dose study of PGN-EDO51 with a long-term extension in participants with duchenne muscular dystrophy amenable to exon 51-skipping treatment (CONNECT1-EDO51) (NCT06079736) 10 Holland A et al.: P27 three novel enhanced delivery oligonucleotide candidates for duchenne muscular dystrophy mediate high levels of exon 53, 45, and 44 skipping. Neuromuscul Disord 2023; 33(Suppl1): 103-4 11 An open-label phase 1b/2 study of WVE-N531 in patients with duchenne muscular dystrophy (NCT04906460) 12 Lai Y et al.: Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J Clin Invest 2009; 119(3): 624-35 13 Vu Hong A et al.: An engineered AAV targeting integrin alpha V beta 6 presents improved myotropism across species. Nat Commun 2024; 15(1): 7965 14 A phase 1/2, multicenter, open-label study to investigate the safety, tolerability, and efficacy of a single intravenous dose of SGT-003 in males with duchenne muscular dystrophy (INSPIRE DUCHENNE) (NCT06138639) 15 Rahm L et al.: Myotonic dystrophy type 1: clinical diversity, molecular insights and therapeutic perspectives. Nat Rev Neurol 2025; 21: 623-41 16 Safety, tolerability, PK, and PD study of PGN-EDODM1 in participants with myotonic dystrophy type 1 (FREEDOM-DM1) (NCT06204809)17 A phase 2 randomized, double-blind, placebo-controlled, multiple ascending dose study of PGN-EDODM1 in adult participants with myotonic dystrophy type 1 (FREEDOM2-DM1) (NCT06667453)
Das könnte Sie auch interessieren:

Modèles prédictifs pour l’indication de la GPE chez les patient·es atteint·es de SLA
La gastrostomie percutanée endoscopique (GPE) est couramment utilisée dans la sclérose latérale amyotrophique (SLA) avancée. Le moment optimal de la mise en place fait toutefois l’objet ...
Mécanismes d’action de l’ECT en tant que traitement alternatif de la dépression
La dépression compte parmi les troubles psychiques les plus fréquents et les plus invalidants, mais ses fondements neurobiologiques ne sont à ce jour que partiellement élucidés. L’ ...

Psychothérapie augmentée à la kétamine
La (S-)kétamine, antidépresseur à action rapide, est utilisée de manière efficace chez les patient·es présentant une résistance aux traitements. Pour les doses subanesthésiques, on ...